网页魔域游戏“氮中心手性”再登Nature,这次是List组的无环N
“氮中心手性”再登Nature,网页魔域游戏这次是List组的无环N-手性胺
前些天,我们刚刚报道了南方科技大学谭斌教授团队与加州大学洛杉矶分校K. N. Houk教授团队合作发表的一篇Nature文章(Nature, 2025, DOI: 10.1038/s41586-025-09607-6, 点击阅读详细),他们通过不对称有机催化控制三角锥形氮中心手性,并高对映选择性地合成了21个N-立体中心为唯一手性元素的1,2-oxazolidines类化合物。值得一提的是,这是N-手性化合物的首例催化不对称合成。 谈到手性,学过中学化学的人一般会想起四面体碳中心及其相关结构的手性分子。目前,基于碳中心的立体化学已得到广泛而深入的研究,但是三角锥形手性分子的研究仍相对不足,目前常见的化学合成仅限于P-手性磷化氢、S-手性亚砜和锍盐等少数体系(图1A)。2023年,Martin D. Smith等人首次报道了桥联、螺旋手性氧鎓离子的不对称合成(Nature, 2023, 615, 430, 点击阅读详细)。相比之下,当手性中心是氮原子时,合成就会变得非常具有挑战性,特别是对于开链胺来说。N-手性叔胺在室温条件下构型不稳定,通常需要通过桥联或季铵化形成相应的四面体铵离子以防止“伞形翻转”引起的外消旋化。近几十年来,化学家在含或不含其它手性元素的N-手性环胺合成方面已取得显著进展,但目前尚未稳定无环N-手性胺的不对称合成案例见诸报道。早在1890年,Hantzsch和Werner等人就预测三价手性胺可能具有光学活性,但后续分离获取均未成功。随后,Meisenheimer等人指出由于手性胺存在快速锥形反转,因此难以拆分。进一步研究发现在氮原子上引入强电负性和Lewis碱性取代基(如氧原子)会显著提高锥形手性分子的翻转能垒。事实上,Kostyanovsk等人早在1980年已成功分离出差向异构胺1a和1b(图1B),并测定其反转能垒与半衰期。 2021年,德国马克斯•普朗克煤炭研究所Benjamin List教授课题组报道了IDPi催化硅基保护的烯酮缩醛(SKA)与原位生成的双硅基硝鎓离子(bis-silylnitronium ions)的不对称加成反应(Nat. Catal., 2021, 4, 1043,点击阅读详细),并成功构建了一系列有价值的脂肪族β3-氨基酸中间体。在此基础上,Benjamin List教授与同事Chandra Kanta De博士等人利用IDPi催化烯醇硅烷与硝鎓离子的不对称加成策略,对映选择性地合成了一系列稳定的无环N-手性胺(图1C)。在所得的无环N-手性胺中,两个N-氧取代基抑制了氮翻转,从而显著减缓异构化。此外,本工作不仅实现了N-手性中心的有效构建,也为深入探索尚未充分开发的手性差向异构胺的化学性质提供了新途径。相关成果发表在Nature 上。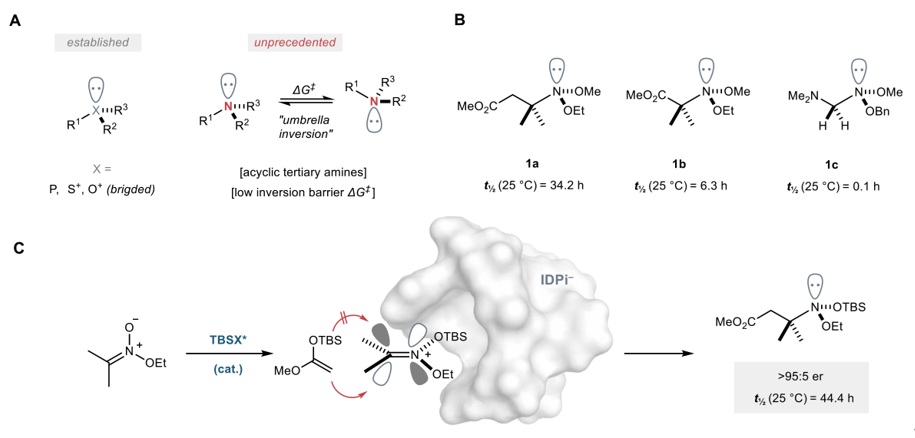
图1. 锥形分子的不对称合成。图片来源:Nature
首先,作者以异丙基nitronate(2a)和烯醇硅烷(3a)为模型底物对一系列强酸性、受限的IDPi Brønsted酸催化剂进行筛选(图2A),结果显示带有强吸电子Tf基团的手性Brønsted酸(4a-4e)能以良好的产率和对映选择性得到产物;而当Tf被五氟磺酰基(C6F5SO2)取代时,催化剂4f-4j的对映选择性更高,其中螺芴基取代的催化剂4f效果最好(产率:95%,e.r.值:90:10)。进一步筛选螺芴7位烷基取代基后,发现催化剂4k能以68%的产率和96.6:3.4 e.r.值得到所需胺1d(图2B),并且测得1d的半衰期为44.4 h、活化自由能(ΔG‡,25°C)为25.2 kcal/mol,特别是胺1d的半衰期高于已报道的异头胺1a(图2C-2D),作者推测这可能是由于硅原子的存在或者其空间位阻大所致。其次,作者探索了该反应的底物范围(图2E),结果表明:1)硝基甲烷衍生的nitronate 2e在4h的催化下以95%的产率(¹H NMR)和92.5:7.5 e.r.值得到所需产物1e,但其在25°C下极不稳定,因此无法准确测定半衰期;2)硝基环己烷衍生的nitronate 2f在优化条件下以良好的产率(64%)和中等的对映选择性(80.7:19.3 e.r.)获得胺1f,但其半衰期仅13.3 h,不足1d的三分之一;3)将SKA中的烷基从乙基改为异丁基或苄基(1g-1i)后,均能以中等至良好的对映选择性获得相应产物且半衰期相近;4)将氮上乙氧基换为甲氧基后得到胺1j(产率:93%,e.r.值:92.8:7.2),而且半衰期显著延长(58.3 h);5)在SKA中引入大位阻TDS基团,使用4h催化能以80%的产率和96.8:3.2 e.r.值获得1k;6)将nitronate的R2烷基替换为另一个硅基时,可以36-47%的产率和67.7:32.3 e.r.-75: 25 e.r.值获得产物1l和1m,并且半衰期极长(分别为256.7 h和288.8 h)。

图2. 反应发展、平衡稳定性研究及氮的取代基效应。图片来源:Nature
接下来,作者对该反应的立体选择性控制进行了研究。先前的研究已证实在SKA与nitronate 反应中会形成硅基硝鎓离子/IDPi反阴离子对。在该反应中,反应性碳原子及其取代基可明确区分Re和Si面。然而,由于硝鎓离子碳上的取代基相同,尽管相应的氮原子具备Re/Si特征,但无法进行形式上的面区分。因此,需采用新的对映选择性描述方式,并且Izumi和Tai先前提出的对映异构体区分(enantiomer differentiation,图3A)、对映面区分(enantiofacial differentiation,图3B)和对映(位)基团区分(enantio(topic)-group differentiation,图3C)均不适用于本体系。受Ioffe工作的启发,作者合成了未取代环状nitronate 2n并对TBSOTf催化2n与SKA 3a的反应进行了研究,结果表明该反应仅生成cis-1n,符合反式共平面加成路径(图3D)。显然,该机制也适用于非环状nitronates 2的反应。在关键的成键步骤中,电子正式从亲核试剂(硅基保护的烯酮缩醛)的π-HOMO转移至亲电试剂(硝鎓离子)的π*-LUMO,而且该π*轨道具有两个节点,C=N-π体系的同相叶瓣位于相对平面。电子密度向其中一个π*叶瓣转移,这不仅启动新C–C-σ键形成,还导致C=N-π键断裂,并在氮原子另一侧积累电子密度,形成孤对电子。因此,碳上两个π*叶瓣的区分决定了氮的绝对构型,此种独特的对映选择性可定义为“叶瓣对映体区分(enantiolobal differentiation)”(图3E)。
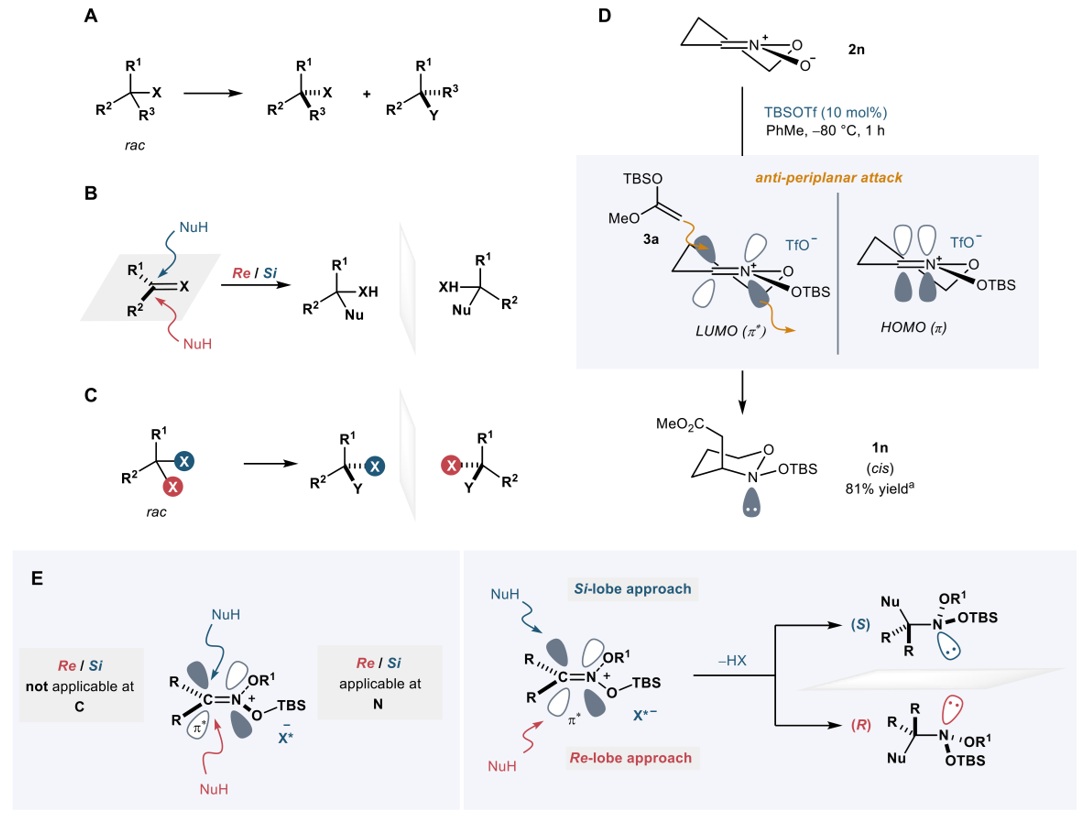
图3. 立体化学研究。图片来源:Nature
最后,作者通过密度泛函理论(DFT)计算对主要对映体(TSmaj)和次要对映体(TSmin)的过渡态进行了研究,以深入了解在IDPi 4j催化体系中关键的对映选择性差异。TSmaj与TSmin的能量差与实验结果定性一致。在两种路径中,SKA加成至硝鎓离子后,氮原子在π平面另一侧积累电子密度并最终形成相应的氮中心手性(图4A)。对映选择性源于精细调控的微环境,其中大位阻硅基与手性抗衡离子之间产生空间位阻(图4B)。在两种情况下,亲核试剂的接近方式都尽量减少了大位阻硅基与手性抗衡离子之间的空间位阻。相比之下,Si-波瓣通过底物与催化活性位点中的Lewis碱性官能团之间的非共价相互作用获得了显著的稳定作用,而Re-波瓣则较少发生此类相互作用,其可能是由于亲核试剂上的硅基朝外定向,这得到了畸变相互作用分析的支持。
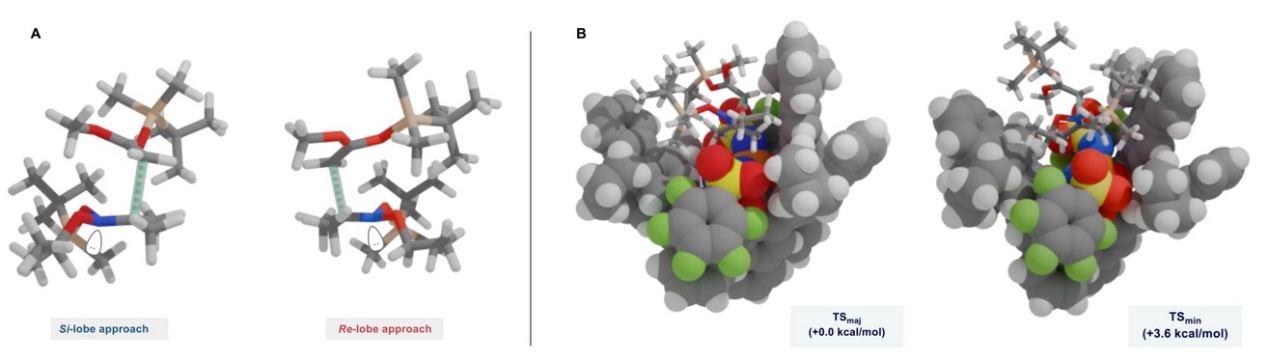
图4. 主要异构体(左)和次要异构体(右)的过渡态。图片来源:Nature
总结
本文利用IDPi催化烯醇硅烷与硝鎓离子的不对称加成策略,对映选择性地合成了一系列稳定的无环N-手性胺。在所得的N-无环手性胺中,两个N-氧取代基抑制氮反转,从而显著减缓异构化。这一策略的成功得益于类酶IDPi催化剂,其可构建明确定义的不对称环境,有效控制锥形氮中心的立体化学。本研究为氮立体控制提供了新范式。作者预期,所提出的核心概念将推动后续研究,促进不对称催化与分子立体化学领域的进一步发展。
原文(扫描或长按二维码,识别后直达原文页面,或点此查看原文):

The Asymmetric Synthesis of an Acyclic N-Stereogenic Amine
Chendan Zhu, Sayantani Das, Marie Sophie Sterling, Nobuya Tsuji, Spencer J. Léger, Frank Neese, Chandra Kanta De, Benjamin List
Nature, 2025, DOI: 10.1038/s41586-025-09905-z
导师介绍
Benjamin List
https://www.x-mol.com/university/faculty/50088
(本文由吡哆醛供稿)
如果篇首注明了授权来源,任何转载需获得来源方的许可!如果篇首未特别注明出处,本文版权属于 X-MOL ( x-mol.com ), 未经许可,谢绝转载!
点击分享
收藏
取消收藏